GLUT1 deficiency syndrome
GLUT1 deficiency syndrome (GLUT1 DS) was first described in 1991. It is one of the few epilepsies in childhood that can be treated effectively by initiating a special “ketogenic” diet. For this reason this condition should be excluded in any child with intractable epilepsy by means of a fasting lumbar puncture.
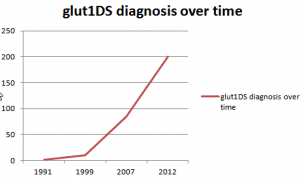
Klepper 2014
there are over 200 known patients worldwide.
Many questions about the course and the multiple manifestations of the disease are still unanswered.
Here are some examples:
* as the number of patients increases, the different forms and patterns of the disease will be better understood,
* Long-term effects, side effects and the best time to stop the ketogenic diet will become clearer
* An animal model (GLUT1-knockout mouse) would provide valuable information about the disease itself and the success of new therapies
* the administration of oral ketones could optimize the ketogenic diet,
* The frequency of the disease in adults is still completely unexplored.
We are just beginning to understand and treat this condition.
reason
The brain needs glucose for energy. This search on the best transporter GLUT1 from the blood into the brain.
If this transporter is defective, there is a deficiency of fuel (hypoglycorrhagia), an "energy crisis" in the brain - just the glucose transporter (GLUT1) defect.
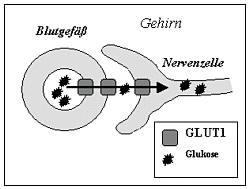
The most common cause of the transporter defect is a mutation in the SLC2A1 gene (OMIM 138140, gene map locus 1p35-31.3)
The phenotype is very variable and several atypical variants have been described. (see symptoms)
symptoms
main symptoms
cerebral, mostly resistant to therapy seizures in the first year of life, (heaped sober); Improvement after glucose intake
Infancy: Nick attacks, eye roiling, complex partial seizures; no BNS attacks
Childhood: slowing down, "how drunk", myoclonic seizures, Grand Mal
Adults: Incident type variable; still little experience
more symptoms are:
• acquired microcephalus
• hypotension
• Ataxia
• dystonia
In addition, the phenotypic manifestation of the Glut1 defect manifests itself in a broad variance that expresses the conflict between movement disorder, cognitive impairments and epilepsy.
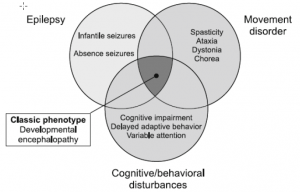
According to literature (1), various movement disorders are observed in patients with Glut1 defect. Such as dystonia, chorea, tremor, dyspraxia and myoclonus. The expression is very individual, but seems to be the more pronounced, the later the Glut1 defect is recognized and adequately treated.
The extended phenotype also differentiates between other types of movement disorders such as paroxysmal or PEDystonia.
The following overview gives an overview of how often various movement disorders have been observed.(1)
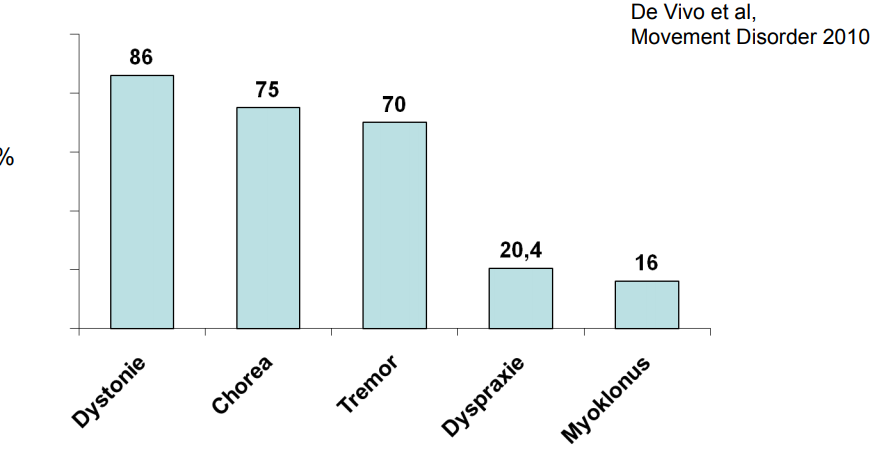
Also in the epileptic manifestation of the Glut1 defect, variable, often therapy-resistant, types of seizures can be observed. As:
- cyanosis-attacks
- absences
- focal / generalized seizures
- Myoclonisch-astatically
In addition, there seems to be an accumulation of epilepsy in the literature in the early childhood of the Glut1 defect.
perspectives
The first patients with GLUT1 deficiency syndrome were described in 1991. Ever since, great progress has been made in understanding this novel entity; but yet, many open questions remain:
* despite increasing numbers of patients identified no classification on clinical or genetic grounds has so far been possible due to the complexity of the disease. Transient GLUT1 deficiency syndrome and patients without seizures have been described; the manifestation of this disease in adults is not yet defined.
*Long-term effects and the time-point when to discontinue the ketogenic diet are currently unclear. Recently a GLUT1-deficient mouse has been generated. Research in this field will help to understand the disease-mechanisms and enable therapeutical trials.
*Oral ketones could possibly be administered in addition to the ketogenic diet, decreasing the fat to non-fat ratio and making the diet more palatable.
We are only beginning to understand and treat this complex novel disease.
diagnostic approach
The biochemical hallmark of GLUT1 deficiency syndrome is a low glucose concentration in the cerebrospinal fluid (CSF), termed “hypoglycorrhachia”.
This is determined in a fasting lumbar puncture. Absolute glucose values in CSF can be misleading as they directly relate to blood glucose concentrations.
For instance, a CSF glucose of 30 mg/dl would be normal if blood glucose is 50 mg/dl (ratio 0,6), but indicative for GLUT1 deficiency syndrome if blood glucose is 100 mg/dl (ratio 0,3). Therefore hypoglycorrhachia is expressed as a ratio of CSF vs. blood glucose concentrations.
A ratio of < 0.4 is indicative for GLUT1 deficiency syndrome.
However, the value does not correlate to the severity of the disease.
Quotient (Example):

Important:
The lumbar puncture has to be performed in afasting state (4 - 6 hours fasting prior to the investigation), as changes in glucose concentrations are reflected much faster in blood than in brain. Blood glucose should be determined before the lumbar puncture to avoid a stress-related elevation of this important parameter.
Always determine lactate in the cerebrospinal fluid (in GLUT1 deficiency syndrome it is always low to normal).
Cerebral imaging (CT, MRI) usually is uninformative. Brain malformations are not a feature of GLUT1 deficiency syndrome. Imaging of the glucose metabolism in the brain (PET) has been reported to show a pattern specific for GLUT1 deficiency syndrome.
If the diagnosis is confirmed by evidence of repeated low CSF glucose and low quotient, the number and function of the GLUT1 transporter in red blood cells can be determined. These elaborate examinations are reserved for special laboratories.
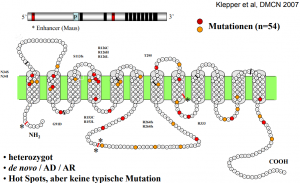
The gene for GLUT1 is known: it is located on the short arm of chromosome 1 (1p35-31.3). It can therefore be examined for mutations. For this purpose, a simple blood collection (EDTA blood) is sufficient - the diagnostics are also carried out in special laboratories.
Meanwhile, many mutations have been detected in patients with GLUT1 defect.
therapy
The ketogenic diet should always be in the hands of an experienced team of physicians and dieticians. It is a very strict and limiting diet – vitamins, minerals and trace elements need to be supplemented and the fat to non-fat ratio needs to be strictly adhered to in every meal.
The response to the ketogenic diet in GLUT1 deficiency syndrome is striking. Almost all patients become seizure-free within weeks without comedication. Patients and parents also report a positive impact of the diet on mental alertness and endurance.
Several drugs (caffeine, phenobarbital, narcotics, chloral-hydrate) inhibit GLUT1 function. Others interact with the ketogenic mechanisms of the diet (valproate, topiramate, acetazolamide). These substances should be avoided in GLUT1 deficient patients.
perspectives
The first patients with GLUT1 deficiency syndrome were described in 1991. Ever since, great progress has been made in understanding this novel entity; but yet, many open questions remain:
* despite increasing numbers of patients identified no classification on clinical or genetic grounds has so far been possible due to the complexity of the disease. Transient GLUT1 deficiency syndrome and patients without seizures have been described; the manifestation of this disease in adults is not yet defined.
* Long-term effects and the time-point when to discontinue the ketogenic diet are currently unclear.
* Recently a GLUT1-deficient mouse has been generated. Research in this field will help to understand the disease-mechanisms and enable therapeutical trials.
* Oral ketones could possibly be administered in addition to the ketogenic diet, decreasing the fat to non-fat ratio and making the diet more palatable.
We are only beginning to understand and treat this complex novel disease.
FAQ